2026
Carbon doping in GeTe drives differences in local structure and properties
J. D. Langhout, M. D. Kelley, D. N. Alverson, L. M. Barnes, C. Mercier, D. Old, D. P. Adams, M. M. Butala. Carbon doping in GeTe drives differences in local structure and properties. J. Chem. Mater. C (2026) asap article.
Abstract
Advances in low-power, energy-efficient information storage and computing require understanding and controlling the atomic and nanoscale structures of functional materials, such as phase-change materials. Phase-change memory technology enables nonvolatile, low-power memory in devices by storing information through reversible changes in a phase-change material's atomic structure (i.e., transformations between amorphous and crystalline phases) that have corresponding changes in properties, including electronic resistivity and optical reflectivity. Here, we apply complementary X-ray absorption spectroscopy and X-ray pair distribution function analyses to experimentally identify the local- and medium-range atomic structure differences of GeTe and C-doped GeTe thin films. Upon controlled heating, composition- and temperature-dependent atomic structure evolution in GeTe and C-doped GeTe films shows differences in bonding behavior and local structure that directly influence crystallization onset temperature. We find that the introduction of C interrupts Ge–Ge bonds in amorphous GeTe, altering the as-deposited structure to be more similar to the distorted rocksalt structure of crystalline α–GeTe. The change alters the response of the amorphous atomic structure to heating and also lowers the crystallization onset temperature, from 230 °C in GeTe to 220 °C in the C-doped film. The combined insights from both X-ray techniques provide understanding of structural transformations that enables the development and optimization of next-generation memory and computing materials.

Predicting Short-Range Order in Disordered Rocksalt Li-Oxide Cathode Materials with Density Functional Theory and Crystal Graph Neural Networks
R. S. Ullberg, J. D. Langhout, M. M. Butala, S. R. Phillpot. Predicting Short-Range Order in Disordered Rocksalt Li-Oxide Cathode Materials with Density Functional Theory and Crystal Graph Neural Networks. ACS Appl. Energy Mater. 9 (2026) 3703-3715.
Abstract
Li-excess disordered rocksalts have recently emerged as a promising class of materials to replace conventional layered nickel manganese cobalt oxide cathodes as a means to enhance the reversible capacity of Li-ion batteries. However, Li ions on the cation sublattice have been shown to form localized clusters of chemical short-range order, which can affect capacity and Li mobility, high values of which are crucial to the macroscale performance of the cathode. To address this challenge, we employ both ab initio calculations and crystal graph machine learning methods to reveal structural factors contributing to short-range order formation and explore a large compositional space to identify materials that exhibit a thermodynamically stable disordered phase. Ultimately, this high-throughput screening process results in a subset of compositions that are strong candidates for future experimental characterization.
Esterification synthesis of iron oxide nanoparticle tracers for magnetic particle imaging (MPI)
A. C. Velazquez-Albino, B. Elsea, A. Melnyk, N. Eswaran, E. D. Imhoff, A. G. Williams, W. Graham, J. A. Johnson, C. E. Johnson, M. M. Butala, C. M. Rinaldi-Ramos. Esterification synthesis of iron oxide nanoparticle tracers for magnetic particle imaging (MPI). Nanoscale, 18 (2026) 2625-2640.
Abstract
The potential for producing iron oxide magnetic particle imaging (MPI) tracers using an alternative synthesis method based on esterification of iron oleate with oleyl alcohol was evaluated. The defined reaction mechanism allows monitoring of reaction progress with Fourier Transform Infrared (FTIR) spectroscopy. The influence of reaction temperature and precursor flow rate on esterification reactions of Fe(ii) oleate for tuning iron oxide nanoparticle size and dispersity was studied, identifying conditions for producing larger nanoparticles suitable as MPI tracers. Increasing temperature and decreasing flow rates were found to increase the resulting nanoparticle size and reduce dispersity. Furthermore, the effect of the iron source used to prepare the iron oleate precursor was evaluated by characterization of nanoparticle magnetic properties, composition, and MPI performance. Although the nature of the precursor did not appear to affect nanoparticle morphology or growth, it influenced magnetic properties and MPI performance. Saturation magnetization was close to the bulk value of magnetite and the discrepancy between physical and magnetic diameters was lowest for nanoparticles synthesized with oleates prepared using Fe(ii) or Fe(iii), as opposed to nanoparticles synthesized using an oleate prepared with a 1 : 2 molar mixture of Fe(ii) and Fe(iii). X-ray diffraction characterized that nanoparticles synthesized using the Fe(iii) oleate are the most crystalline, followed by Fe(ii) and the 1 : 2 Mix, respectively. Mössbauer spectroscopy was used to verify iron oxide phases, suggesting nanoparticles synthesized using the Fe(iii) oleate consist of a mixture of γ-Fe2O3 and Fe3O4, in contrast to those obtained from the Fe(ii) and 1 : 2 Mix oleates, which consisted of a mixture of wüstite, γ-Fe2O3 and Fe3O4. Characterization of MPI performance using a MOMENTUM™ scanner demonstrated the capability of the esterification reaction to yield high-quality monodisperse MPI tracers.

2025
Distinguishing isotropic and anisotropic signals for X-ray total scattering using machine learning
D. N. Alverson, D. Olds, M. M. Butala*
Distinguishing isotropic and anisotropic signals for X-ray total scattering using machine learning. Acta. Cryst. (2025). A81, 175-187.
Abstract
Understanding structure–property relationships is essential for advancing technologies based on thin films. X-ray pair distribution function (PDF) analysis can access relevant atomic structure details spanning local-, mid- and long-range structure. While X-ray PDF has been adapted for thin films on amorphous substrates, measurements on single-crystal substrates are necessary to accurately determine structure origins for some thin film materials, especially those for which the substrate changes the accessible structure and properties. However, when measuring films on single-crystal substrates, high-intensity anisotropic Bragg spots saturate 2D detector images, overshadowing the thin films' isotropic scattering signal. This renders previous data processing methods for films on amorphous substrates unsuitable for films on single-crystal substrates. To address this measurement need, we developed IsoDAT2D, an innovative data processing approach using unsupervised machine learning algorithms. The program combines dimensionality reduction and clustering algorithms to separate thin film and single-crystal substrate X-ray scattering signals. We use SimDAT2D, a program we developed to generate simulated thin film data, to validate IsoDAT2D. We also use IsoDAT2D to isolate X-ray total scattering signal from a thin film on a single-crystal substrate. The resulting PDF data are compared with similar data processed using previous methods, especially substrate subtraction for single-crystal and amorphous substrates. PDF data from IsoDAT2D-identified X-ray total scattering data are significantly better than from single-crystal substrate subtraction, but not as reliable as PDF data from amorphous substrate subtraction. With IsoDAT2D, there are new opportunities to expand PDF to a wider variety of thin films, including those on single-crystal substrates, with which new structure–property relationships can be elucidated to enable fundamental understanding and technological advances.

Mo-substitution in V2O5 tunes the structure towards three-dimensional connectivity and improves Li-ion battery cycling
K. Parui, B. G. Gardner, K. A. Pitton, N. G. Caracuel, E. Vidal-Torres, S. Gandhi, B. S. Guiton, J. C. Nino, M. M. Butala.* Mo-substitution in V2O5 tunes the structure towards three-dimensional connectivity and improves Li-ion battery cycling. J. Phys. Mater. (2025). 8 025017.
Abstract
The development of alternative energy sources is crucial for reducing reliance on fossil fuels, particularly for mobile applications such as personal electronics and transportation. This necessitates the advancement of battery materials based on abundant and inexpensive constituent elements. To achieve this requires investigating materials in a broader compositional and structural design space. Early transition metal oxides, including the intercalation electrode V2O5, however, the performance of V2O5 is hindered by phase transformations during battery cycling that lead to capacity fade and short device lifetimes. This study investigates the modification of V2O5 through Mo substitution in a series of the form VMoxO5 for x = 0.05, 0.1, 0.2, 0.4, 0.6, and 0.8. X-ray diffraction data reveal progressive structural changes with increasing Mo content, which in turn change the progression of phase transformations during the first discharge. The different product also results in different cycling profile shapes that indicate differences in the charge storage mechanism as a function of Mo content. As a result, samples with higher Mo-substitution, especially V1.2Mo0.8O5, have narrower hysteresis, higher capacity, and improved capacity retention. While there is a limited solubility of Mo in the V2O5 structure, with secondary phases and defects at many compositions, we show that Mo substitution alters the cycling behavior of V2O5 to deep discharge, which can inform the design of intercalation materials for energy storage applications.

2024
Isovalent substitution modulates average and short-range structure in disordered rocksalt oxides
J. D. Langhout, E. Gager, T. Ulloa, S. Shepard, J. C. Nino, and M. M. Butala.*
Isovalent substitution modulates average and short-range structure in disordered rocksalt oxides. J. Mater. Chem. A, 12 (2024) 32140-32153.
Abstract
Li-excess disordered rocksalt oxides are promising candidate materials for high-energy density Li-ion battery cathodes. Their disordered cation sublattice provides opportunity to design compositions that balance performance and sustainability, especially enabling the use of abundant and inexpensive elements. However, relationships between composition, short-range cation ordering, and their effects on performance are not well-understood. Here, we use a compositional series of the form Li1.2Mn0.4Ti0.4−xZrxO2 , in which Ti4+ is gradually replaced with Zr4+ , to study the effect of Zr content on average- and short-range structure using synchrotron X-ray diffraction and pair distribution function analysis. We report the coexistence of multiple modes of short-range order, which have a major impact on battery capacity. However, the effects of Zr on degree of short-range ordering, lattice parameter, and chemical segregation also influence battery capacity, reflecting the complex dependencies of composition on structure across length scales in these disordered materials.

Cross-linking organic cathodes enhances stability at the expense of ionic accessibility
A. Davis,♦ K. Parui,♦ A. M. M. Hasan, L. A. Pineda, J. D. Langhout, K. A. Treaster, M. M. Butala,* A. M. Evans.* Cross-linking organic cathodes enhances stability at the expense of ionic accessibility. J. Mater. Chem. A, 12 (2024) 28874-28881. (♦ equal contribution)
Abstract
We have investigated the fundamental impacts of network cross-linking density on organic cathode performance. Current battery technologies rely on transition metal oxide cathodes that suffer from significant availability, cost, environmental, and humanitarian drawbacks. This reality has inspired the exploration of organic cathode materials that host high theoretical capacities, are environmentally friendly, and are tunable by molecular design. These desirable features are typically accompanied by undesirable instability during battery cycling due to solubility in electrolyte, leading to diminished capacities. Cross-linking polymer electrodes presents one strategy to address dissolution challenges. Here, we synthesized variably cross-linked naphthalene diimide (NDI)-based networks to systematically study the effect of cross-linking density on organic electrode battery performance. NDI-networks were cast as composite electrodes, manufactured into coin cells with lithium metal anodes, and evaluated by galvanostatic cycling. We observed that increased cross-linking facilitated reversible redox-access to NDI units in the network, which correlated to increased stability and capacities. Cathodes with optimized cross-linking host an initial capacity of 106 mA h g−1 and >75% capacity retention after 100 cycles with a C/10 rate. This contrasts with uncross-linked materials, which rapidly diminished in performance due to dissolution in the electrolyte, and more densely cross-linked materials, which suffered from limited ionic accessibility. These findings demonstrate that (1) cross-linking can improve organic electrode performance and (2) there are tradeoffs between cycling stability, capacity, and overall energy storage performance with cross-linking density. Future investigations will explore how network design and processing conditions can be leveraged to co-optimize organic cathode performance across these important metrics.

Magnetic structure and properties of the compositionally complex perovskite (Y0.2La0.2Pr0.2Nd0.2Tb0.2)MnO3
N. D. Arndt,* B. L. Musico, K. Parui, K. Sahebkar, Q. Zhang, A. R. Mazza, M. M. Butala, V. Keppens, T. Z. Ward, and R. F. Need.* Magnetic structure and properties of the compositionally complex perovskite (Y0.2La0.2Pr0.2Nd0.2Tb0.2)MnO3. J. Mater. Chem. C, 12 (2024) 13474-13484.
Abstract
Large configurational disorder in compositionally complex ceramics can lead to unique functional properties that deviate from traditional rules of alloy mixing. In recent years, compositionally complex oxides (CCOs) have shown intriguing magnetic behavior including long-range order, enhanced magnetic exchange couplings, and mixed phase magnetic structures. This work focuses on how large local spin disorder affects magnetic ordering in a CCO. Specifically, we investigated the A-site alloyed perovskite, (Y0.2La0.2Pr0.2Nd0.2Tb0.2)MnO3, or (5A)MnO3, using a combination of bulk magnetometry, synchrotron X-ray diffraction, and temperature-dependent neutron diffraction. The five A-site ions have an average spin and ionic radius nearly equal to that of Nd3+ ions, which minimizes structural distortions and allows for an understanding of the local spin disorder effects through a direct comparison with NdMnO3. Our magnetometry data show that (5A)MnO3 exhibits two distinct phase transitions associated with the A-site and B-site sublattices, as seen in NdMnO3, as well as the presence of domain pinning and exchange bias at low temperature, suggesting a mixed phase magnetic ground state, as seen in other magnetic CCOs. Neutron powder diffraction shows clear long-range antiferromagnetic ordering below 67 K and refines to a Pn′ma′ magnetic structure at low temperature, in excellent agreement with the well-studied behavior of NdMnO3. The two most notable differences in (5A)MnO3 magnetism apparent from our data are a slight suppression of the B-site ordering temperature, which is explained by a smaller Mn–O–Mn bond angle in (5A)MnO3 than NdMnO3, and the presence of a magnetic susceptibility transition above the B-site ordering, which could indicate the formation of a cluster glass but requires further study. This work demonstrates a general method of isolated investigation of size and spin disorder in CCOs and motivates future work using local structure probes to better understand the effects of nanoscale clustering and local spin disorder in magnetic CCOs.

Multifunctional COF design addresses Li-S organic electrode limitations
K. A. Treaster, A. N. Davis, M. M. Butala, and A. M. Evans.* Multifunctional COF design addresses Li-S organic electrode limitations. Trends in Chemistry, 6 (2024) P503-505.
Abstract
Lithium-sulfur (Li-S) batteries are restricted by cathode polysulfide shuttling and anode lithium dendrite formation. Jin, Zuo, and coworkers recently showed that Li-S batteries with high capacities and cycling stabilities emerge from intentionally designed covalent organic framework (COF) electrodes. This report highlights how COF design can address fundamental challenges in organic electrode engineering.
Local structure effects of carbon-doping on the phase change material Ge2Sb2Te5
J. D. Langhout, D. N. Alverson, C. Ginter, B. Ravel, D. P. Adams, M. M. Butala.*
Local structure effects of carbon-doping on the phase change material Ge2Sb2Te5. J. Mater. Chem. C, 12 (2024) 7867-7877.
Abstract
Ge2Sb2Te5 is used in phase change memory, a nonvolatile memory technology, due to its phase change properties. The primary advantage of phase change memory over the state-of-the-art (flash memory) is its simple and small device geometry, which allows for denser nodes and lower power consumption. In phase change memory, resistive heating induces fast switching between the high resistance amorphous and low resistance crystalline phases, corresponding to storage of low and high digital states, respectively. However, the instability of the amorphous phase of Ge2Sb2Te5 presents issues with processing and long-term data storage; such issues can be resolved by C doping, which stabilizes the amorphous phase and raises the crystallization temperature. To better understand the local structural effects of C doping on Ge2Sb2Te5, in situ Ge K-edge X-ray absorption spectroscopy measurements were taken during heating of films with various C doping concentrations. The range of structural transformation temperatures derived from X-ray absorption near-edge structure analysis across the C doping series proved narrower than crystallization temperatures reported in similar in situ X-ray diffraction experiments, which may reflect changes in local structure that precede long-range ordering during crystallization. In addition, rigorous extended X-ray absorption fine structure fitting across and between temperature series revealed effects of C doping on the rigidity of Ge–Te bonds at low (2 at% and 4 at%) C concentrations.
Supramolecular design as a route to high-performing organic electrodes
A. N. Davis, K. Parui, M. M. Butala, and A. M. Evans. Supramolecular design as a route to high-performing organic electrodes. Nanoscale, 16 (2024) 10142-10154.

2023
Phosphate-functionalized Zirconium Metal–Organic Frameworks for Enhancing Lithium–Sulfur Battery Cycling
B. Liu, A. E. Baumann, M. M. Butala, V. S. Thoi, Phosphate-functionalized Zirconium Metal–Organic Frameworks for Enhancing Lithium–Sulfur Battery Cycling. Chem. Eur. J. 2023, 29, e202300821.
Abstract
Lithium–sulfur batteries are promising candidates for next-generation energy storage devices due to their outstanding theoretical energy density. However, they suffer from low sulfur utilization and poor cyclability, greatly limiting their practical implementation. Herein, we adopted a phosphate-functionalized zirconium metal–organic framework (Zr-MOF) as a sulfur host. With their porous structure, remarkable electrochemical stability, and synthetic versatility, Zr-MOFs present great potential in preventing soluble polysulfides from leaching. Phosphate groups were introduced to the framework post-synthetically since they have shown a strong affinity towards lithium polysulfides and an ability to facilitate Li ion transport. The successful incorporation of phosphate in MOF-808 was demonstrated by a series of techniques including infrared spectroscopy, solid-state nuclear magnetic resonance spectroscopy, and X-ray pair distribution function analysis. When employed in batteries, phosphate-functionalized Zr-MOF (MOF-808-PO4) exhibits significantly enhanced sulfur utilization and ion diffusion compared to the parent framework, leading to higher capacity and rate capability. The improved capacity retention and inhibited self-discharge rate also demonstrate effective polysulfide encapsulation utilizing MOF-808-PO4. Furthermore, we explored their potential towards high-density batteries by examining the cycling performance at various sulfur loadings. Our approach to correlate structure with function using hybrid inorganic–organic materials offers new chemical design strategies for advancing battery materials.
R-Nb2O5 has an ‘idealized’ V2O5 structure and Wadsley-Roth-like structural stability during Li-ion battery cycling
K. Parui, A. Lee, S. Gandhi, and M. M. Butala.* R-Nb2O5 has an ‘idealized’ V2O5 structure and Wadsley-Roth-like structural stability during Li-ion battery cycling. J. Mater. Chem. A, 11 (2023) 5559-5567.
Abstract
The adoption of batteries across diverse applications requires electrode materials with a wider range of performance metrics, such as cost, safety, and material availability. Along the path to discovering new commercially viable materials, a fundamental understanding of chemical and atomic structure features that provide structural stability and effective ion transport is essential. In support of new understanding, we report the cycling behavior of metastable R-Nb2O5. R-Nb2O5 adopts an ‘idealized’ V2O5 structure, in which [NbO6] octahedra alternate in edge- and corner-sharing resulting in ReO3-like slabs, whereas Wadsley–Roth materials have ReO3-like blocks, linked through edge-sharing octahedra at intersecting crystallographic shear planes. We find that this slab structure is stable during cycling, with minor atomic structure changes and cycling curves that are symmetric on discharge and charge, resembling the behavior of Wadsley–Roth materials more than other related materials, such as ReO3, V2O5, or Nb3O7F. Based on our findings, R-Nb2O5 can serve as a ‘structural bridge’ between Wadsley–Roth block structures and V2O5, through which we can relate inter- and intra-polyhedral structures to cycling behavior and structural stability during cycling.

2022
Crystal Structure of the tau11-Al4Fe1.7Si
B. Rijal, S. Soto-Medina, K. Parui, A. Sachdev, M. M. Butala, M. V. Manuel, and R. Hennig. Crystal Structure of the tau11-Al4Fe1.7Si. J. Alloys Compd. 902 (2022) 163141.
Abstract
The intermetallic τ11 Al4Fe1.7Si phase is of interest for high-temperature structural application due to its combination of low density and high strength. We determine the crystal structure of the τ11 phase through a combination of powder neutron diffraction and density functional theory calculations. Using Pawley and Rietveld refinements of the neutron diffraction data provides an initial crystal structure model. Since Al and Si have nearly identical neutron scattering lengths, we use density-functional calculations to determine their preferred site occupations. The τ11 phase exhibits a hexagonal crystal structure with space group P63/mmc and lattice parameters of a = 7.478 Å and c = 7.472 Å. The structure comprises five Wyckoff positions; Al occupies the 6h and 12k sites, Fe the 2a and 6h sites, and Si the 2a sites. We observe site disorder and partial occupancies on all sites with a large fraction of 80% Fe vacancies on the 2d sites, indicating an entropic stabilization of the τ11 phase at high temperature.

2021
Energy Storage mechanisms in vacancy-ordered Wadsley-Roth layered niobates
K. McColl, K. J. Griffith, R. L. Dally, R. Li, J. E. Douglas, K. R. Poeppelmeier, F. Cora, I. Levin, and M. M. Butala. Energy Storage mechanisms in vacancy-ordered Wadsley-Roth layered niobates. J. Mater. Chem. A 9 (2021) 20006-20023. Invited, Emerging Investigator Issue
Abstract
Wadsley–Roth (WR) crystallographic shear structures demonstrate high energy and power densities as Li-ion battery anode materials. We report the (de)lithiation behavior of two WR-derived layered niobates: NaNb3O8 and KNb3O8. Both demonstrate multi-electron (Nb5+/Nb3+) redox on the first discharge, reacting with ≈5 mol Li per mol ANb3O8. Li intercalation in NaNb3O8 is dominated by Li-diffusion kinetics and evolution of the interlayer structure...
2019
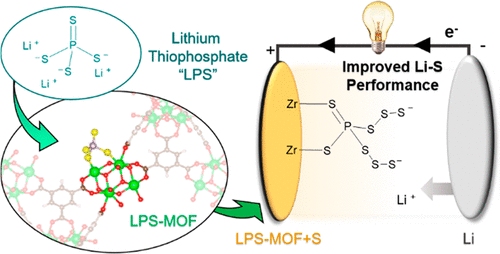
Lithium Thiophosphate Functionalized Zirconium MOFs for Li-S Batteries with Enhanced Rate Capabilities
A. E. Baumann, X. Han, M. M. Butala, and V. S. Thoi. Lithium Thiophosphate Functionalized Zirconium MOFs for Li-S Batteries with Enhanced Rate Capabilities. J. Am. Chem. Soc. 141 (2019) 17891–19899
Abstract
Zirconium metal–organic frameworks (Zr-MOFs) are renowned for their extraordinary stability and versatile chemical tunability. Several Zr-MOFs demonstrate a tolerance for missing linker defects, which create “open sites” that can be used to bind guest molecules on the node cluster. Herein, we strategically utilize these sites to stabilize reactive lithium thiophosphate (Li3PS4) within the porous framework for targeted application in lithium–sulfur (Li–S) batteries. Successful functionalization of the Zr-MOF with PS43– is confirmed by an array of techniques including NMR, XPS, and Raman spectroscopy, X-ray pair distribution function analysis, and various elemental analyses. During electrochemical cycling, we find that even a low incorporation extent of lithium thiophosphate in Zr-MOFs improves sulfur utilization and polysulfide encapsulation to deliver a sustainably high capacity over prolonged cycling. The functionalized MOF additives also prevent cell damage under abusive cycling conditions and recover high capacities when the cell is returned to lower charge/discharge rates, imperative for future energy storage devices. Our unique approach marries the promising chemical attributes of the purely inorganic Li3PS4 with the stability and high surface area of MOFs, creating a Li–S cathode architecture with a performance beyond the sum of its component parts. More broadly, this novel functionalization strategy opens new avenues for facile syntheses of “designer materials” where chemical components from discrete disciplines can be united and tailored for specific applications.
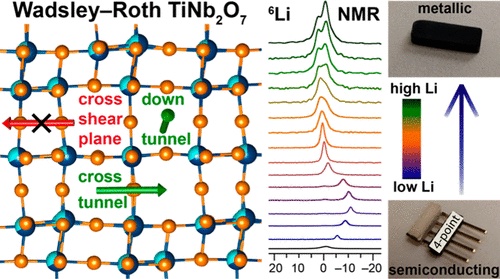
Ionic and Electronic Conduction in TiNb2O7
K. J. Griffith, I. D. Seymour, M. A. Hope, M. M. Butala, L. K. Lamontagne, M. B. Preefer, C. P. Kocer, G. Henkelman, A. J. Morris, M. J. Cliffe, S. E. Dutton, and C. P. Grey. Ionic and Electronic Conduction in TiNb2O7. J. Am. Chem. Soc. 141 (2019) 16706–16725
Abstract
TiNb2O7 is a Wadsley–Roth phase with a crystallographic shear structure and is a promising candidate for high-rate lithium ion energy storage. The fundamental aspects of the lithium insertion mechanism and conduction in TiNb2O7, however, are not well-characterized. Herein, experimental and computational insights are combined to understand the inherent properties of bulk TiNb2O7. The results show an increase in electronic conductivity of seven orders of magnitude upon lithiation and indicate that electrons exhibit both localized and delocalized character, with a maximum Curie constant and Li NMR paramagnetic shift near a composition of Li0.60TiNb2O7. Square-planar or distorted-five-coordinate lithium sites are calculated to invert between thermodynamic minima or transition states. Lithium diffusion in the single-redox region (i.e., x ≤ 3 in LixTiNb2O7) is rapid with low activation barriers from NMR and DLi = 10–11 m2 s–1 at the temperature of the observed T1 minima of 525–650 K for x ≥ 0.75. DFT calculations predict that ionic diffusion, like electronic conduction, is anisotropic with activation barriers for lithium hopping of 100–200 meV down the tunnels but ca. 700–1000 meV across the blocks. Lithium mobility is hindered in the multiredox region (i.e., x > 3 in LixTiNb2O7), related to a transition from interstitial-mediated to vacancy-mediated diffusion. Overall, lithium insertion leads to effective n-type self-doping of TiNb2O7 and high-rate conduction, while ionic motion is eventually hindered at high lithiation. Transition-state searching with beyond Li chemistries (Na+, K+, Mg2+) in TiNb2O7 reveals high diffusion barriers of 1–3 eV, indicating that this structure is specifically suited to Li+ mobility.
2018
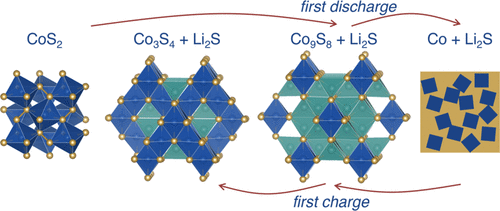
Operando Studies Reveal Structural Evolution with Electrochemical Cycling for the Li–CoS2 System
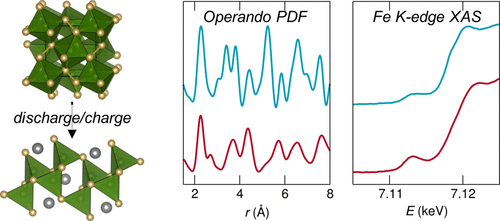
M. M. Butala, V. V. T. Doan-Nguyen, A. J. Lehner, C. Göbel, M. A. Lumley, S. Arnon, K. M. Wiaderek, O. J. Borkiewicz, K. W. Chapman, P. J. Chupas, M. Balasubramanian, and R. Seshadri. Operando Studies Reveal Structural Evolution with Electrochemical Cycling for the Li–CoS2 System. J. Phys. Chem. C, 122 (2018) 24559–24569
Abstract
The drive toward high energy density alternatives to Li-ion batteries has led to great interest in energy storage materials not inherently constrained by the capacity limits of the currently employed intercalation electrode materials. Among the alternatives under consideration are electrode materials with theoretical capacities many times greater than intercalation electrodes that store charge through so-called conversion reactions. However, the significant structural changes that enable the high theoretical capacity of conversion systems contribute to issues of poor efficiency and short cycle life. To better understand cycling issues in conversion systems, we study the local structure evolution of CoS2 during Li storage. Being metallic and potentially capable of redox on both anion and cation sites, CoS2 would be expected to display promise as a cathode material. Through combined ex situ X-ray absorption near-edge spectroscopy and pair distribution function analysis from operando X-ray total scattering, we describe the reactions that take place over the first 1.5 cycles. In doing so, we identify the irreversible formation of a Co9S8-like local structure with significantly limited electrochemical activity as the primary source of capacity fade. The methods employed here and the insights that emerge could inform the rational design of conversion systems for electrochemical energy storage.
2017
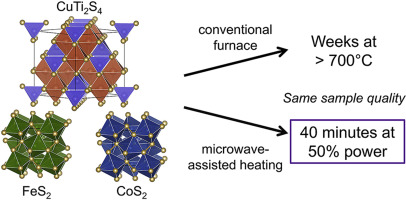
Rapid Microwave-Assisted Preparation of Binary and Ternary Transition Metal Sulfide Compounds
M. M. Butala, M. A. Perez, S. Arnon, C. Göbel, M. B. Preefer, and R. Seshadri. Rapid Microwave-Assisted Preparation of Binary and Ternary Transition Metal Sulfide Compounds. Solid State Sciences, 74 (2017) 8–12
Abstract
Transition metal chalcogenides are of interest for energy applications, including energy generation in photoelectrochemical cells and as electrodes for next-generation electrochemical energy storage. Synthetic routes for such chalcogenides typically involve extended heating at elevated temperatures for multiple weeks. We demonstrate here the feasibility of rapidly preparing select sulfide compounds in a matter of minutes, rather than weeks, using microwave-assisted heating in domestic microwaves. We report the preparations of phase pure FeS2, CoS2, and solid solutions thereof from the elements with only 40 min of heating. Conventional furnace and rapid microwave preparations of CuTi2S4 both result in a majority of the targeted phase, even with the significantly shorter heating time of 40 min for microwave methods relative to 12 days using a conventional furnace. The preparations we describe for these compounds can be extended to related structures and chemistries and thus enable rapid screening of the properties and performance of various compositions of interest for electronic, optical, and electrochemical applications.
Local Structure Evolution and Modes of Charge Storage in Secondary Li–FeS2 Cells
M. M. Butala, M. Mayo, V. V. T. Doan-Nguyen, M. A. Lumley, C. Göbel, K. M. Wiaderek, O. J. Borkiewicz, K. W. Chapman, P. J. Chupas, M. Balasubramanian, G. Laurita, S. Britto, A. J. Morris, C. P. Grey, and R. Seshadri. Local Structure Evolution and Modes of Charge Storage in Secondary Li–FeS2 Cells. Chem. Mater., 29 (2017) 3070–3082
Abstract
In the pursuit of high-capacity electrochemical energy storage, a promising domain of research involves conversion reaction schemes, wherein electrode materials are fully transformed during charge and discharge. There are, however, numerous difficulties in realizing theoretical capacity and high rate capability in many conversion schemes. Here we employ operando studies to understand the conversion material FeS2, focusing on the local structure evolution of this relatively reversible material. X-ray absorption spectroscopy, pair distribution function analysis, and first-principles calculations of intermediate structures shed light on the mechanism of charge storage in the Li–FeS2 system, with some general principles emerging for charge storage in chalcogenide materials. Focusing on second and later charge/discharge cycles, we find small, disordered domains that locally resemble Fe and Li2S at the end of the first discharge. Upon charge, this is converted to a Li–Fe–S composition whose local structure reveals tetrahedrally coordinated Fe. With continued charge, this ternary composition displays insertion–extraction behavior at higher potentials and lower Li content. The finding of hybrid modes of charge storage, rather than simple conversion, points to the important role of intermediates that appear to store charge by mechanisms that more closely resemble intercalation.
2016
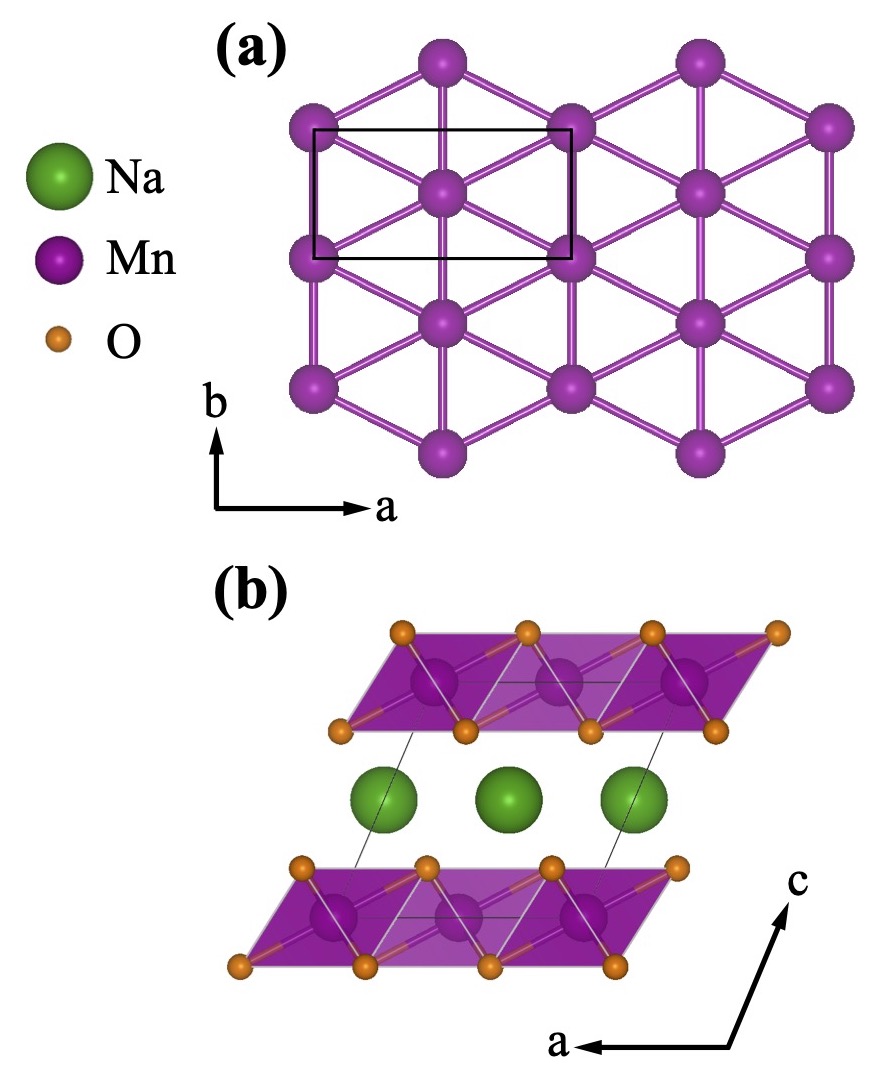
Floating zone growth of α–Na0.9 MnO2 single crystals
R. Dally, R. J. Clément, R. Chisnell, S. Taylor, M. M. Butala, V. V. T. Doan-Nguyen, J. W. Lynn, C. P. Grey, S. D. Wilson. Floating zone growth of α–Na0.9 MnO2 single crystals. J. Cryst. Growth, 459 (2016) 203–208
Abstract
Single crystal growth of α-NaxMnO2 (x=0.90) is reported via the floating zone technique. The conditions required for stable growth and intergrowth-free crystals are described along with the results of trials under alternate growth atmospheres. Chemical and structural characterizations of the resulting α-Na0.90MnO2 crystals are performed using ICP-AES NMR, XANES, XPS, and neutron diffraction measurements. As a layered transition metal oxide with large ionic mobility and strong correlation effects, α-NaxMnO2 is of interest to many communities, and the implications of large volume, high purity, single crystal growth are discussed.
Molybdenum polysulfide chalcogels as high-capacity, anion-redox-driven electrode materials for Li-ion batteries
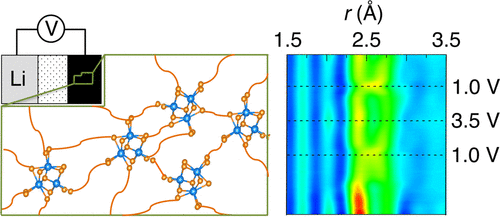
V. V. T. Doan-Nguyen, K. S. Subrahmanyam, M. M. Butala, J. C. Gerbec, S. M. Islam, K. N. Kanipe, C. E. Wilson, M. Balasubramanian, K. M. Wiaderek, O. J. Borkiewicz, K. W. Chapman, P. J. Chupas, M. Moskovits, B. S. Dunn, M. G. Kanatzidis, and R. Seshadri. Molybdenum polysulfide chalcogels as high-capacity, anion-redox-driven electrode materials for Li-ion batteries Chem. Mater., 28 (2016) 8357–8365
Abstract
Sulfur cathodes in conversion reaction batteries offer high gravimetric capacity but suffer from parasitic polysulfide shuttling. We demonstrate here that transition metal chalcogels of approximate formula MoS3.4 achieve a high gravimetric capacity close to 600 mAh g–1 (close to 1000 mAh g–1 on a sulfur basis) as electrode materials for lithium-ion batteries. Transition metal chalcogels are amorphous and comprise polysulfide chains connected by inorganic linkers. The linkers appear to act as a “glue” in the electrode to prevent polysulfide shuttling. The Mo chalcogels function as electrodes in carbonate- and ether-based electrolytes, which further provides evidence of polysulfide solubility not being a limiting issue. We employ X-ray spectroscopy and operando pair distribution function techniques to elucidate the structural evolution of the electrode. Raman and X-ray photoelectron spectroscopy track the chemical moieties that arise during the anion-redox-driven processes. We find the redox state of Mo remains unchanged across the electrochemical cycling and, correspondingly, the redox is anion-driven.
Microplasma spray method for direct, substrate-independent deposition of nanostructured metal oxides
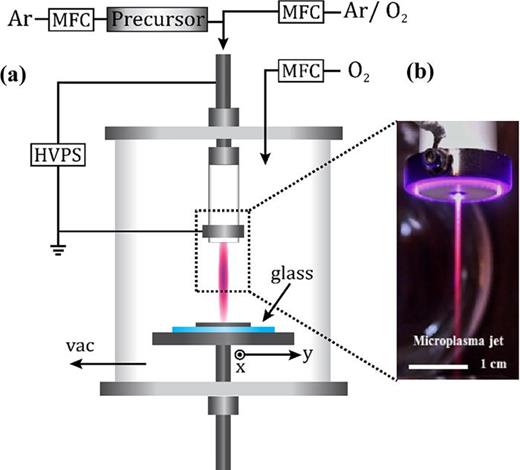
K. E. Mackie, A. C. Pebley, M. M. Butala, J. Zhang, G. D. Stucky, and M. J. Gordon. Microplasma spray method for direct, substrate-independent deposition of nanostructured metal oxides, Appl. Phys. Lett., 109 (2016) 033110
Abstract
A general, substrate-independent method for plasma deposition of nanostructured, crystalline metal oxides is presented. The technique uses a flow-through, micro-hollow cathode plasma discharge (supersonic microplasma jet) with a “remote” ring anode to deliver a highly directed flux of growth species to the substrate. A diverse range of nanostructured materials (e.g., CuO, α-Fe2O3, and NiO) can be deposited on any room temperature surface, e.g., conductors, insulators, plastics, fibers, and patterned surfaces, in a conformal fashion. The effects of deposition conditions, substrate type, and patterning on film morphology, nanostructure, and surface coverage are highlighted. The synthesis approach presented herein provides a general and tunable method to deposit a variety of functional and hierarchical metal oxide materials on many different surfaces. High surface area, conversion-type CuO electrodes for Li-ion batteries are demonstrated as a proof-of-concept example.
MnO conversion in Li-ion batteries: In situ studies and the role of mesostructuring
M. M. Butala, K. R. Danks, M. A. Lumley, S. Zhou, B. C. Melot, and R. Seshadri. MnO conversion in Li-ion batteries: In situ studies and the role of mesostructuring, ACS App. Mater. Interfaces, 8 (2016) 6496–6503
Abstract
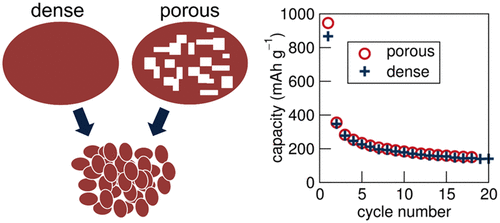
Complex manganese oxides have been extensively studied as intercalation Li-ion battery electrodes. The simple oxide MnO has been proposed as a conversion anode material with a theoretical capacity of 756 mAh g–1 for full reduction to the metal. We report the reaction of MnO with Li using in situ X-ray diffraction and find no sign of crystalline products upon either discharge or charge. However, the absence of reflections, paired with electrochemical impedance spectroscopy, suggests disordered discharge products. We also examine composite electrodes with porous particles of MnO as the active component, with pores generated through the reductive heating of Mn3O4. We compare the behavior of these with more dense MnO powders, including studies of the electrode morphologies pre- and postcyling. We find differences in the first discharge relevant to the utility of such mesostructuring in conversion reaction materials. Specifically, we find this type of mesostructure, which gives advantage in intercalation and pseudocapacitive storage, does not yield the same benefits for conversion reaction systems.
2014
Mesoporous materials from template-free vapor-phase reductive leaching of Zn from Zn—M—O compounds (M = Nb, Mo, W)
C. Lermer,* M. M. Butala,* B. R. Lettiere, and R. Seshadri, Mesoporous materials from template-free vapor-phase reductive leaching of Zn from Zn—M—O compounds (M = Nb, Mo, W), Cryst. Growth Des., 14 (2014) 4526–4530. (*equal contribution)
Abstract
Vapor-phase leaching of Zn and O from complex oxides was performed with the goal of creating mesoporous metal oxides with connected porosity. At elevated temperatures, complex Zn–M–O oxides (M = Nb, Mo, W) can be reduced to yield textured product materials, including reduced M–O oxides, nitrides, and the metal. The nature of the product varies with temperature, time, reducing atmosphere, and the identity of the metal M. M = Nb results in the formation of porous NbO2 without the need for extraneous templates or pore formers. The crystal chemistry of the starting Nb compound is found to influence the nature of the texturing or porosity of the final product. The evolution of morphology is also impacted by the starting Zn:Nb ratio. In the case of the Mo and W compounds, reductive leaching yields the metal or metal nitride (for M = Mo). Morphology change is also observed, and varies for each product phase. An additional interesting aspect of the process is that the reductive leaching occurs in stages, allowing intermediate Zn–M–O compositions with reduced M to be stabilized. The evolution of morphology also appears to be dependent on the initial and final crystal structures.
